Hello! In the bio and medical industries, the clinical trial is an absolute must-have milestone.
While both pathways share the ultimate goal of improving patient health and safety, their research designs and the specific data required by regulatory agencies differ completely. Failing to grasp these fundamental differences can lead to confusion during the protocol design phase, costing you valuable time and resources.
Today, we will break down the core differences and essential regulatory points—from research design to safety evaluation, ethical review, and data management—so you can apply them directly to your practice!
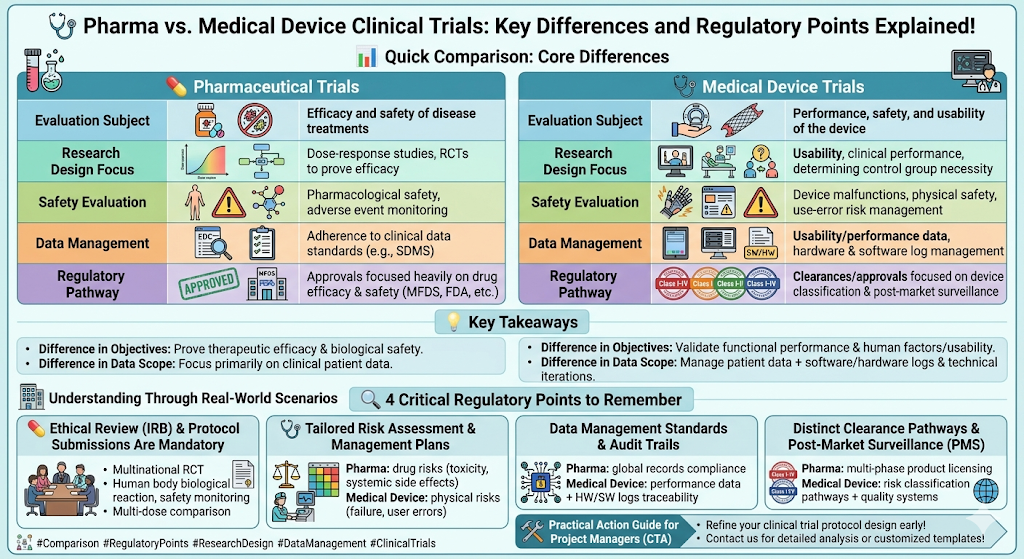
📊 Quick Comparison: Core Differences
Take a direct look at how these two pathways diverge across key parameters:
| Category | 💊 Pharmaceutical Trials | 🩺 Medical Device Trials |
| Evaluation Subject | Efficacy and safety of disease treatments | Performance, safety, and usability of the device |
| Research Design Focus | Dose-response studies, RCTs to prove efficacy | Usability, clinical performance, determining control group necessity |
| Safety Evaluation | Pharmacological safety, adverse event monitoring | Device malfunctions, physical safety, use-error risk management |
| Data Management | Adherence to clinical data standards (e.g., SDMS) | Usability/performance data, hardware & software log management |
| Regulatory Pathway | Approvals focused heavily on drug efficacy & safety (MFDS, FDA, etc.) | Clearances/approvals focused on device classification & post-market surveillance |
💡 Key Takeaways
- Difference in Objectives: Pharmaceuticals must prove ‘therapeutic efficacy’ in treating diseases, whereas medical devices must validate ‘device performance, safety, and human factors/usability.’
- Difference in Data Scope: Pharmaceutical trials focus primarily on clinical patient data. In contrast, medical device trials must manage patient data alongside software update histories, hardware logs, and technical iterations.
🏢 Understanding Through Real-World Scenarios
💊 Case 1: Pharmaceutical Trial (New Drug Development)
In a multinational Phase 3 trial for a new drug, investigators utilize randomized controlled trials (RCTs) to compare efficacy and adverse effects across various dosages. Because the study tracks how the human body biologically reacts to a chemical substance, rigorous ethical reviews, advanced data management systems, and intensive safety monitoring are mandatory.
🩺 Case 2: Medical Device Trial (Diagnostic Equipment)
A multi-center study is launched to evaluate the performance of a new hospital diagnostic device. Since no drug is administered, the focus shifts away from biological side effects. Instead, the trial evaluates whether the device operates without errors in a real-world hospital environment and gathers clinician feedback on usability. Tracking software patch histories and firmware updates during the trial is also critical.
🔍 4 Critical Regulatory Points to Remember
To secure seamless regulatory approval, verify these four essential compliance areas based on your product type:
- Ethical Review (IRB) & Protocol Submissions Are Mandatory
- For both sectors, obtaining Institutional Review Board (IRB) approval and submitting a comprehensive clinical trial protocol is an absolute, non-negotiable requirement to ensure bioethics and participant safety.
- Tailored Risk Assessment & Management Plans
- Pharmaceuticals: Focus heavily on drug-related risks, such as toxicity, systemic side effects, and long-term adverse events.
- Medical Devices: Focus on physical, mechanical, and environmental risks, including device failures, malfunctions, or user operational errors.
- Data Management Standards & Audit Trails
- Pharmaceuticals: Must strictly comply with standardized clinical data management systems and global electronic record regulations.
- Medical Devices: Require robust traceability that accounts for real-world performance data as well as hardware/software logs to maintain a transparent audit trail.
- Distinct Clearance Pathways & Post-Market Surveillance (PMS)
- Pharmaceuticals typically undergo a rigid, multi-phase product licensing process to enter the market. Medical devices follow customized pathways based on their risk classification (Class I–IV) and rely heavily on manufacturer-responsible quality management systems and post-market compliance.
🎯 Final Thoughts & Next Steps
If pharmaceutical trials are all about “proving therapeutic effect and biological safety,” medical device trials focus squarely on “validating functional performance and user adaptation.” While strict data integrity is a shared foundation, the scope of data required by regulators varies greatly. Aligning your strategy early prevents costly operational detours.
🛠️ Practical Action Guide for Project Managers (CTA)
Which pathway does your current project fall under? Use the comparison checklist above to refine your clinical trial protocol design early on.
If you need a detailed case analysis or an optimized clinical trial protocol template tailored to your pipeline, feel free to send us an inquiry! We are here to help clarify your regulatory roadmap.
#Comparison #RegulatoryPoints #ResearchDesign #DataManagement #ClinicalTrials