In the bio and healthcare industries, clinical trials represent the ultimate hurdle to commercialization. However, many people mistakenly think, “I’ve run a pharmaceutical trial, so medical devices should be pretty much the same,” only to face unexpected regulatory hurdles or design flaws that derail their project timelines.
Pharmaceuticals and medical devices have fundamentally different regulatory requirements and design philosophies from inception. In this post, we will clearly break down the core differences between the two sectors and share an easy-to-read comparative checklist and real-world case examples you can apply directly to your next project.
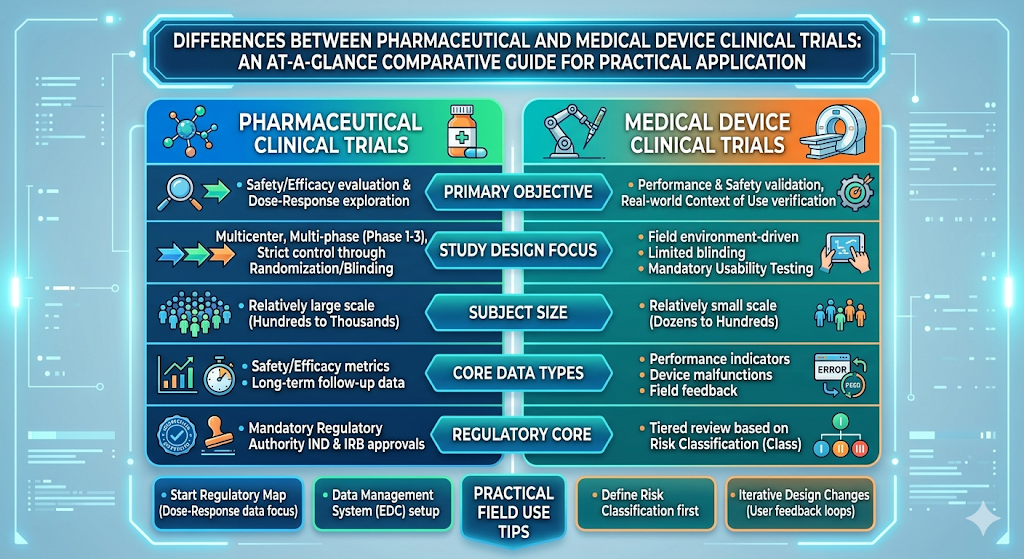
📌 Core Comparison Points
1. Study Design and Subjects
- Pharmaceuticals: These follow a traditional, multi-phase structure (Phase 1 to Phase 3). To evaluate safety and efficacy, studies primarily focus on ‘Dose-Response’ relationships, requiring hundreds to thousands of subjects. Randomization and double-blind designs are essential to establish statistical significance.
- Medical Devices: Rather than sequential phases, the focus is squarely on verifying performance and safety within the actual Context of Use. Due to the nature of physical devices, it is often impossible to blind the investigator or patient. Subject sizes are relatively small, and Usability Testing—evaluating the human-factors engineering of how the device is handled—plays a major role.
2. Regulatory Pathways and Review Procedures
- Pharmaceuticals: Strict approval from regulatory authorities (such as the Ministry of Food and Drug Safety, FDA, etc.) and an Institutional Review Board (IRB) is mandatory for an Investigational New Drug (IND) application. Accumulated Safety Profiles serve as the definitive baseline for approval decisions.
- Medical Devices: The necessity and scope of a clinical trial depend entirely on the device’s Risk Classification (Class I, II, III). If you can prove Substantial Equivalence to an existing predicate, clinical trials may be exempted entirely. Therefore, meticulous documentation and evidence generation that reflect real-world clinical environments are crucial.
3. Data Management and Safety
- Pharmaceuticals: Reporting mechanisms for Serious Adverse Events (SAEs) are extraordinarily stringent, and long-term follow-up tracking is mandatory. The validation requirements for Electronic Data Capture (EDC) systems are also exceptionally high.
- Medical Devices: Unlike drugs, the primary data sources extend beyond side effects to include ‘Device Malfunctions’, usability issues, and user feedback from the field. Change management following device upgrades and field-centric data collection are heavily emphasized.
📊 At-a-Glance Comparison Table
| Category | Pharmaceutical Clinical Trials | Medical Device Clinical Trials |
| Primary Objective | Safety/Efficacy evaluation & Dose-Response exploration | Performance & Safety validation, Real-world Context of Use verification |
| Study Design Focus | Multicenter, Multi-phase (Phase 1-3), Strict control through Randomization/Blinding | Field environment-driven, Limited blinding, Mandatory Usability Testing |
| Subject Size | Relatively large scale (Hundreds to Thousands) | Relatively small scale (Dozens to Hundreds) |
| Core Data Types | Safety/Efficacy metrics, Long-term follow-up data | Performance indicators, Device malfunctions, Field feedback |
| Regulatory Core | Mandatory Regulatory Authority IND & IRB approvals | Tiered review based on Risk Classification (Class) |
💡 Practical Case Studies & Implementation Tips
Case 1: Multi-phase Trial of a New Drug Candidate (Pharmaceutical Workflow)
After establishing a safety profile in the early stage (Phase 1), the sponsor progressively accumulates generalizable efficacy data through multi-national, multi-center studies. Project managers must set up robust Electronic Data Capture (EDC) systems and SAE reporting infrastructures right from the start.
Case 2: Field Evaluation of a New Surgical Device (Medical Device Workflow)
The team first identifies the risk classification of the developed device to confirm whether a clinical trial is mandatory. During the trial, performance indicators are gathered within the context of actual use by medical professionals. Timelines must account for iterative ‘Design Changes’ to the prototype based on real-time user feedback.
🛠 Quick Tips for Practitioners!
- Map out your ‘Regulatory Map’ during the project kickoff. A strategy that secures dose-response data for drugs, or risk class-specific performance/usability data for devices, prevents costly trial-and-error with your budget and timeline.
- Understand the distinct languages of submission documents. If the core language for pharmaceutical documents is “statistical significance and safety profiles,” the core language for medical devices is “performance and risk management tied to a Quality Management System (QMS).”
🎯 Conclusion
Pharmaceutical and medical device clinical trials differ in the fundamental nature of the data they target and how regulatory bodies view them. Recognizing the distinction between the strict, control-oriented design of drug trials and the real-world, context-oriented design of device trials is the first step toward building a successful roadmap.
Use this guide as a foundation during your initial planning phase to establish a data strategy tailored perfectly to your product.
💬 What are your experiences in the field?
If you have faced unexpected hurdles coordinating with regulatory authorities or have questions while preparing for your drug or device clinical trial, feel free to share them in the comments below! If you need a more detailed practical checklist file, subscribe and leave an inquiry to receive our latest updates.