The success or failure of a clinical trial is determined at the very first step: Protocol Design. Vague research questions, unrealistic subject selection criteria, and ambiguous endpoints inevitably bounce back as a boomerang of trial delays and budget spikes.
To create a protocol that is smoothly “executable” in the field while satisfying the strict guidelines of regulatory agencies (FDA, EMA, MFDS, etc.), what strategies are required? Here are practical tips and essential frameworks to dramatically reduce initial design errors.
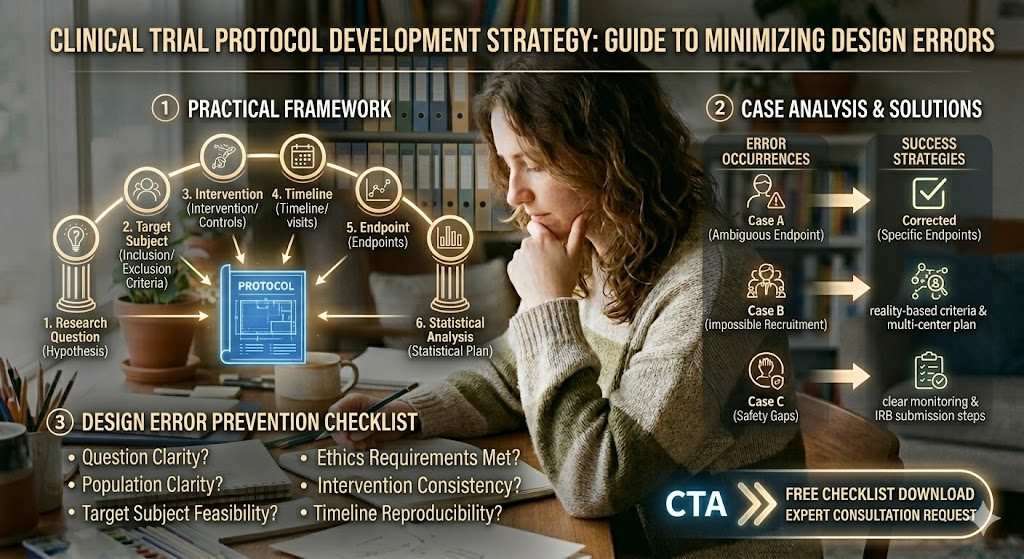
1. Practical Framework for Error-Free Protocol Design
When designing a protocol, you need a systematic reference point to avoid losing your way amid complex timelines and variables. The core framework must be strengthened around the following key elements:
- Research Question: Is the hypothesis and problem definition sharp and clearly verifiable?
- Target Subject: Are the inclusion/exclusion criteria realistic and feasible for recruitment?
- Intervention & Controls: Are the dosage, administration schedule, and control group definitions specific?
- Timeline & Visits: Is the overall trial schedule, subject visit windows, and interim assessment timing seamlessly structured?
- Endpoints: Are the measurement tools for the primary and secondary endpoints objective and reliable?
- Statistical Analysis: Is the justification for the sample size (number of subjects) statistically sound?
- Ethics Requirements: Are the plans for IRB/IEC approval, informed consent forms (ICF), and safety monitoring (DSMB, etc.) fully integrated?
💡 Core Protocol Elements & Checkpoints at a Glance
| Category | Key Content | Critical Practical Warning |
| Inclusion/Exclusion | Disease characteristics, age range, comorbidities | Criteria that are too strict make recruitment impossible, while criteria that are too loose create data bias. |
| Intervention/Controls | Dosage range, route of administration, control definitions | Guidelines must be clear to prevent the mixing of prohibited concomitant medications or interventions in the field. |
| Timeline/Visits | Visit windows, evaluation time points | Schedules must be realistic for both patients and Clinical Research Coordinators (CRCs) to minimize drop-out rates. |
| Endpoints | Primary and secondary endpoints | Objective indicators and validated measurement tools (scales) recognized by regulatory agencies must be utilized. |
2. Case Analysis: Design Errors vs. Solutions
Here are three of the most frequent error cases occurring in actual clinical fields, along with the real strategies used to correct them.
❌ Case A: Ambiguous Endpoint Leading to Altered Analysis Plans
- The Issue: The measurement timing and criteria for the primary endpoint were vague, forcing a modification of the Statistical Analysis Plan (SAP) right before the Data Lock ➡️ Resulted in severe trial delays.
- 💡 Success Strategy: The primary endpoint was specifically defined at the early stage of protocol development, and potential conflicts with secondary endpoints were simulated and preemptively addressed in the protocol.
❌ Case B: Impossible Recruitment Due to Unrealistic Criteria
- The Issue: The conditions for eligible patients (inclusion criteria) were set too strictly, failing to reach the target sample size ➡️ Inability to complete the trial within the designated timeline.
- 💡 Success Strategy: Re-evaluated the feasibility of inclusion/exclusion criteria based on actual prescription patterns and patient pool data, and swiftly transitioned from a single-center to a multi-center recruitment plan.
❌ Case C: Delayed IRB Approval Due to Safety Gaps
- The Issue: Adverse Event (AE/SAE) reporting procedures and data safety monitoring plans were non-specific ➡️ Resulted in consecutive IRB deferrals and delayed interim reviews.
- 💡 Success Strategy: Supplemented the protocol with clear, step-by-step monitoring procedures and Data Safety Monitoring Board (DSMB) operational guidelines to secure swift approval.
3. Design Error Prevention Checklist
Before signing off on your final draft, see if you can confidently answer “YES” to all 7 questions below:
- [ ] Q1. Question Clarity: Is the research question (hypothesis) clear and formulated in a statistically verifiable way?
- [ ] Q2. Population Clarity: Are the inclusion/exclusion criteria realistic for patient recruitment while minimizing bias?
- [ ] Q3. Intervention Consistency: Do the intervention conditions and control settings maintain a consistent standard?
- [ ] Q4. Timeline Reproducibility: Are the patient visit windows and evaluation schedules reproducible in actual clinical settings?
- [ ] Q5. Endpoint Reliability: Is the validity and reliability of the tools measuring the primary endpoint objectively secured?
- [ ] Q6. Statistical Feasibility: Does the statistical plan align with the research question, and is the sample size calculation justified?
- [ ] Q7. Ethics Requirements Met: Are ethical and regulatory requirements—such as informed consent, safety monitoring, and IRB submission steps—completely fulfilled?
🎯 Conclusion: It’s Time to Review Your Protocol
A robustly designed protocol is your strongest shield against unpredictable risks. If you have a draft protocol on your desk right now, look back at the “Target Subject Feasibility” and “Endpoint Clarity” before anything else.
If you want to eliminate errors from the start and perfectly reflect regulatory requirements, undergoing an expert screening is the safest and fastest route.